

O nemoci

RS a jiná demyelinizační onemocnění CNS
1/ Relaps-remitentní forma RS
Je nejčastější a takto probíhá až u 85 % pacientů. Jednotlivé ataky – relapsy – se objevují v průběhu času (týdnů, měsíců, let) a léčbou nebo spontánně se upraví do původního stavu uzdravení – remise. Mezi jednotlivými atakami může být i několik let, ale může se objevit i 1–2× ročně. Asi u 3 % pacientů probíhá tzv. progredující relabující forma RS, kdy po atakách nedojde k úpravě obtíží do původního stavu, což po každé atace vede k nárůstu pacientova hendikepu a mezi atakami se pomalu zhoršuje neurologické postižení.
2/ Sekundárně progresivní RS
Prezentuje se menším počtem atak a vede k pomalému a postupnému zhoršování postižení. Do této fáze se bez včas zavedené imunomodulační léčby dostane po 10–20 letech více než polovina diagnostikovaných pacientů s relaps-remitentní formou RS. Jen velmi malému počtu pacientů je sekundárně progresivní RS diagnostikována bez předchozí relaps-remitentní formy.
3/ Primárně progresivní forma RS
Vyskytuje se asi u 10 % pacientů, začíná v pozdějším věku (okolo 40 let) a častější je u mužů. Při tomto průběhu se neobjevují ataky a neurologické potíže se zhoršují od samotného začátku. Tito pacienti jsou hybně limitovaní podstatně dříve než u předchozí formy.
V roce 2014 proběhla revize klasifikace RS, kdy mezinárodní komise navrhla, aby byla forma onemocnění upřesněna diagnostickými výstupy z MR klasifikace onemocnění na základě jeho aktivity (relaps-remitentní forma s aktivitou × relaps-remitentní forma bez aktivity, progresivní forma s aktivitou × progresivní forma bez aktivity). Zároveň bylo navrženo, aby byl klinicky izolovaný syndrom (CIS, clinically isolated syndrome, tedy první příznaky RS bez jednoznačného potvrzení diagnózy RS pomocnými vyšetřovacími metodami) zařazen jako typ RS.
Průběh onemocnění lze dělit na maligní a benigní. Maligní průběh RS je charakterizován těžkými atakami s rychlým nárůstem invalidity a postupující atrofií mozku na MR. Pacient již zhruba do 5 let maligního průběhu potřebuje vozík. Jako benigní se dá RS označit až zpětně po mnoha letech nemoci, kdy po této době mají pacienti minimální omezení a nízký počet atak.
Začátky účinné léčby RS lze datovat do šedesátých let 20. století, kdy bylo zjištěno, že léčba kortikoidy vede k rychlejší úpravě stavu při akutních atakách. Další zlom nastal v roce 1993, kdy se na trh dostal první chorobu modifikující lék (DMD), interferon beta. Od té doby se léčebná paleta stále rozšiřuje a v současnosti je pro pacienty s relabující RS k dispozici více než desítka DMD s různými mechanismy účinku. Jejich cílem je minimalizovat zánětlivou aktivitu v CNS, tedy v ideálním případě zamezit výskytu / omezit závažnost relapsů, zabránit aktivitě pozorované na magnetické rezonanci a co nejvíce omezit progresi a zhoršování neurologického deficitu. Kromě konkrétního typu DMD je klíčové i načasování terapie a vzájemná posloupnost léčby. Jednoznačně platí, že čím dříve je léčba zahájena, tím větší je její efekt a menší míra postižení v čase. V současnosti dostupné DMD ovlivňují pouze zánětlivou složku, která je dominantní v počátečních fázích nemoci. Zánět je však odpovědný za následnou neurodegeneraci (zánik nervových vláken a buněk). Terapie schopná snížit nebo zvrátit postižení pomocí opravných mechanismů včetně remyelinizace či obnovy nervových vláken je zatím bohužel pouze nedosaženým ideálem.
DMD jsou nasazovány v rámci dvou základních terapeutických strategií:
1/ eskalační léčba – zpočátku jsou nasazovány základní DMD pro RS s nižší aktivitou nemoci (interferon beta, glatirameracetát, teriflunomid a dimethylfumarát) a teprve při nedostatečné účinnosti je léčba eskalována na HET (high efficacy treatment – léky pro RS s vyšší aktivitou nemoci),
2/ časná intenzivní strategie s nasazením HET již od počátku nemoci.
Farmakoterapeutická paleta roztroušené sklerózy je široká a možnosti stále přibývají. Stejně tak se rozšiřují znalosti etiopatogeneze a zdokonaluje se sledování či predikce průběhu nemoci. Pro úspěšnost terapie je však zásadní včasná a správná diagnostika a vedení léčby neurologem se zkušenostmi s touto problematikou. Neméně důležitý je komplexní přístup k pacientovi a mezioborová spolupráce, zvláště s psychoterapeutem a fyzioterapeutem.
Rozhodnutí o léčbě se odehrává v centrech vysoce specializované péče pro RS a NMOSD, kterých je v ČR 15.
Je třeba zdůraznit, že nedílnou součástí léčby RS jsou změny životního stylu v zájmu co nejlepšího zdraví mozku (doporučení Brain Health in MS).
Centra specializované péče pro pacienty s roztroušenou sklerózou a neuromyelitis optica v ČR:
Všeobecná fakultní nemocnice v Praze, U Nemocnice 499/2, 128 08 Praha 2
Fakultní nemocnice v Motole, V Úvalu 84, 150 06 Praha 5
Thomayerova nemocnice, Vídeňská 800, 140 59 Praha 4 – Krč
Fakultní nemocnice Královské Vinohrady, Šrobárova 1150/50, 100 34 Praha 10
Fakultní nemocnice Hradec Králové, Sokolská 581, 500 05 Hradec Králové
Fakultní nemocnice Olomouc, I. P. Pavlova 185/6, 779 00 Olomouc
Fakultní nemocnice Ostrava, 17. listopadu 1790, 708 52 Ostrava – Poruba
Fakultní nemocnice Plzeň, Edvarda Beneše 1128/13, 301 00 Plzeň
Fakultní nemocnice u sv. Anny v Brně, Pekařská 664/53, 656 91 Brno
Krajská zdravotní, a. s. – Nemocnice Teplice, o. z. Duchcovská 53, 415 29 Teplice
Nemocnice České Budějovice, a. s., Boženy Němcové, 585/54, 370 01 České Budějovice
Nemocnice Jihlava, příspěvková organizace, Vrchlického 4630/59, 586 33 Jihlava
Nemocnice Pardubického kraje, a. s., Kyjevská 44, 532 03 Pardubice
Fakultní nemocnice Brno, Jihlavská 340/20, 625 00 Brno
Krajská nemocnice Tomáše Bati, a. s., Havlíčkovo nábřeží 600, 760 01 Zlín
Neuromyelitis optica a onemocnění jejího širšího spektra (NMO spectrum disorders, NMOSD) je zánětlivé onemocnění CNS, dříve často zaměňované za RS, které se však liší imunopatologickými procesy týkajícími se akvaporinových kanálů a klinickým průběhem. Zánětlivě jsou postiženy především zrakové nervy a mícha. Ataky NMOSD jsou převážně klinicky závažné a pacienta ohrožují reziduální invaliditou až smrtí. Onemocnění začíná většinou kolem 40. roku života a 5–10× častěji postihuje ženy. Jeho prevalence se v ČR odhaduje na 1 : 100 000 obyvatel. Vzhledem k tomu, že u mnoha pacientů se vyvinuly ataky zánětu zrakového nervu a míchy v různém odstupu nebo měli projevy onemocnění i mimo optikospinální lokalizaci (mozkový kmen, diencefalon a další), byl implementován termín „neuromyelitis optica spectrum disorders“ – NMOSD. Zahrnuje především tzv. časté klíčové klinické příznaky (optická neuritida, myelitida, syndrom area postrema) a tzv. méně časté klinické příznaky (různé kmenové příznaky, projevy postižení diencefalonu a další).
V akutním stavu se léčebně zasahuje kortikosteroidy, eventuálně výměnnou plazmaferézou, dlouhodobě se u pacientů s přítomností protilátek proti akvaporinu 4 používá satralizumab, inebilizumab, ekulizumab, popř. ravulizumab, u pacientů s nepřítomností protilátek proti akvaporinu 4 imunosuprese azathioprinem, protilátkou proti CD20 (např. rituximab), cyklofosfamidem, mitoxantronem, mykofenolát mofetilem, intravenózními imunoglobuliny, metotrexátem, často v kombinaci s orálními steroidy. Jde o vzácné, ale velmi závažné onemocnění s prognózou těžké invalidity, mnoho pacientů je léta vedeno pod diagnózou roztroušené sklerózy a účinné léky jsou podány pozdě.
Péče o pacienty s RS a NMOSD může být zlepšena včasnou diagnostikou a včasným zahájením imunomodulační léčby. Její efektivita i vedlejší účinky musí být sledovány a léčba v případě nedostatečného účinku včas změněna. Péče o tyto pacienty vyžaduje multidisciplinární tým, a je proto soustředěna do specializovaných center.
Onemocnění s pozitivitou protilátek proti myelinovému oligodendrocytárnímu glykoproteinu (MOGAD, myelin oligodendrocyte glycoprotein antibody-associated disease) je relativně nová diagnostická jednotka, která se vyčlenila ze spektra onemocnění neuromyelitis optica (NMOSD), dříve nazývaného také morbus Devic. Toto zánětlivé autoimunitní demyelinizační onemocnění postihuje zrakový nerv, míchu a některé další struktury centrální nervové soustavy. Jedním z kardinálních projevů u dospělých pacientů je optická neuritida. U dětí se onemocnění manifestuje jako akutní demyelinizační encefalomyelitida. Hlavní diagnostická kritéria jsou patrné známky demyelinizace CNS a průkaz sérových protilátek MOG‑IgG. Akutní léčba spočívá v intravenózní aplikaci steroidů a výměně plazmy.
Autoimunitní encefalitidy (AIE) jsou vzácná akutní či subakutní monofázická nebo progresivní zánětlivá onemocnění CNS podmíněná autoimunitními mechanismy (působením patogenních autoprotilátek nebo autoagresivních efektorových buněk). Na rozdíl od demyelinizačních onemocnění dominuje u AIE postižení korové. Klinický obraz a prognóza se u jednotlivých syndromů velmi liší. Zásadní roli v diagnostice AIE má vyšetření specifických protilátek. Léčbu AIE lze rozdělit na symptomatickou terapii, imunoterapii a onkologickou léčbu (u paraneoplastických syndromů).

Migréna a jiné bolesti hlavy
Mezinárodní klasifikace bolestí hlavy (ICHD-3) dělí bolesti hlavy do 4 skupin: I. primární bolesti hlavy; II. sekundární bolesti hlavy; III. neuropatie, obličejové bolesti a jiné bolesti hlavy a IV. apendix.
Primární bolesti hlavy, mezi které patří migréna, tenzní typ bolesti hlavy, trigeminové autonomní bolesti hlavy a další primární bolesti hlavy, nemají známý organický podklad, který je možno zobrazit pomocí CT či MRI nebo detekovat jinou metodou. Samotným problémem je zde právě bolest. Primární bolesti hlavy se proto klasifikují podle jejich projevů. Etiologický princip zde nelze uplatnit, protože mechanismus jejich vzniku dosud zůstává na úrovni jen částečně ověřených teorií.
Ve skupině sekundárních bolestí hlavy je bolest projevem organického
onemocnění. Jde o velice různorodou skupinu bolestí hlavy jak po stránce lokalizace a intenzity,
tak frekvence a závažnosti, od naprosto banální příčiny až po nejzávažnější, někdy
i fatálně končící stavy.
Příčinou sekundární bolesti hlavy může být úraz nebo cévní onemocnění hlavy a/nebo krku, intrakraniální onemocnění, farmakologicky účinná látka nebo její vysazení, infekce, poruchy hemostázy, postižení lebky, krku, očí, uší, nosu, paranazálních dutin, zubů, dutiny ústní nebo jiných obličejových či krčních struktur nebo psychiatrická porucha.
Celkově výrazně převažují benigní primární bolesti hlavy, se stoupajícím věkem narůstá výskyt sekundárních bolestí. Základní diagnostická rozvaha by vždy měla směřovat k rozlišení mezi primární a sekundární bolestí hlavy. Při diferenciální diagnóze je nejdůležitější pečlivé odebrání anamnézy a neurologické vyšetření. Zjišťuje se charakter bolesti (tupá, pulzující, šlehavá), lokalizace bolesti (hemikranie, difuzní, za okem), její intenzita, délka trvání (sekundy, hodiny, dny, každodenní bolest), četnost výskytu bolesti (několikrát denně, měsíčně), další doprovodné příznaky (fonofobie, fotofobie, odorofobie, lakrimace, sekrece z nosu, paréza mozkových nervů, hemiparéza, mozečkové příznaky, porucha vědomí) a vyvolávající faktory (fyzická aktivita, defekace, předklon hlavy, stres, menstruace, léky, alkohol).
Správně stanovená diagnóza, prevence a léčba primárních bolestí hlavy dokážou v současné době výrazně zlepšit kvalitu života pacientů. Paleta léčiv je široká, od slabších analgetik, protizánětlivých a ergotaminových přípravků, sumatriptanu a jeho derivátů, anxiolytik a myorelaxancií až po antidepresiva.
U sekundárních bolestí hlavy se jejich další průběh odvíjí od základního onemocnění, které je jejich příčinou.
Migréna
Migréna je dnes chápána jako komplexní vrozená cyklická porucha funkce mozku. V cyklickém průběhu nemoci se rozlišuje pět fází: fáze prodromální, aura, bolest hlavy, fáze postdromální a interiktální.
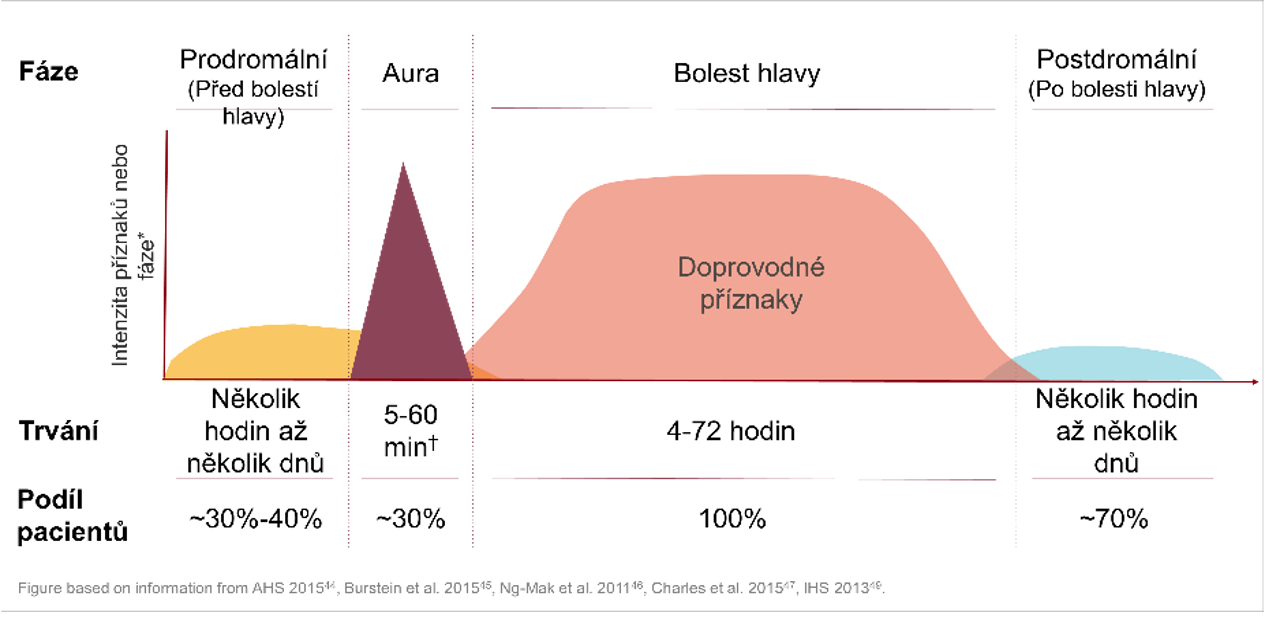
Tyto fáze na sebe mohou chronologicky navazovat, mohou se však i částečně překrývat anebo se u některých pacientů některé z těchto fází vůbec nemusí vyskytnout. Komplikací migrény je status migrenosus, kdy záchvat bolesti trvá déle než 72 hodin, vzácně migrenózní infarkt, perzistující aura bez infarktu a migrénou spuštěný epileptický záchvat.
Onemocnění charakterizují ataky bolestí hlavy, mezi nimiž je pacient bez obtíží. Migrenózní bolest hlavy je popisována jako středně těžká až velmi intenzivní, pulzujícího nebo tepavého charakteru, vyskytující se temporálně a frontálně, typicky unilaterální, přičemž se strany u jednoho pacienta mohou mezi atakami střídat. Často jsou bolesti doprovázeny nauzeou, někdy i se zvracením.
Migréna je diagnostikována na základě podrobné anamnézy, klinického obrazu a negativního výsledku vyšetřovacích metod. V případě pochybností nebo v rámci širší diagnostické rozvahy je vždy vhodné provést zobrazení CNS.
Léčba migrény
K léčbě migrenózní ataky se podle intenzity bolesti a tolerance používají jednoduchá analgetika, nesteroidní antiflogistika a specifická protimigrenózní analgetika – triptany. Opioidní analgetika se v léčbě bolestí hlavy nedoporučují. V klasické profylaktické terapii se používají preparáty z různých lékových skupin, nejčastěji antiepileptika. Při výběru je nutné zohlednit komorbidity pacienta. Aktuální trend v terapii migrény spočívá v cílení na jeden z mechanismů etiopatogeneze rozvoje migrenózní ataky. Jedná se o CGRP (calcitonine gene-related peptide), který se nachází v periferním i centrálním nervovém systému a po aktivaci CGRP receptoru způsobuje vazodilataci, neurogenní zánět a periferní senzitivizaci v trigeminovaskulárním komplexu a tím rozvoj migrenózní ataky. V posledních letech byly vyvinuty monoklonální protilátky s afinitou k CGRP nebo jeho receptoru, které jsou schopny zablokovat aktivaci tohoto komplexu, a tedy i rozvoj bolestí hlavy. Registrovány byly celkem čtyři přípravky, v České republice jsou dostupné zatím jen tři z nich. Úhrada této finančně náročné léčby ze zdravotního pojištění je vázána na centra pro diagnostiku a léčbu bolestí hlavy a splnění definovaných podmínek. Další novinkou jsou gepanty, malé molekuly zaměřené také proti CGRP. Podle farmakokinetických vlastností mohou jednotlivé molekuly gepantů sloužit jako akutní terapie i jako profylaxe.

Alzheimerova choroba a demence
Alzheimerova nemoc (AN) je nejčastější příčinou demence, která stojí za 50 až 60 % případů. Způsobuje typický obraz poruchy epizodické paměti a dalších kognitivních funkcí s postupnou ztrátou („progresivní amnestická demence“). Základními neuropatologickými nálezy jsou atrofie mozku (nejvýraznější mediální temporální struktury), zánik především hipokampálních a cerebrokortikálních neuronů, ztráta synapsí, tvorba beta-amyloidu, formace amyloidových plaků, amyloidová angiopatie, degenerace tau-proteinu, tvorba tzv. tangles (neurofibrilární klubka), neuronální zánětlivé změny v mozku a nadměrná neuronální apoptóza.
Nemoc obvykle začíná zcela nenápadně a rozvíjí se pomalu a postupně po dobu několika let. V průběhu onemocnění se mohou objevit změny osobnosti a chování. U mnoha pacientů je navíc pozorován neklid, agresivita a/nebo deprese. Kromě toho se zhoršuje schopnost úsudku a vyjadřovacích schopností. Navzdory úpadku duševních schopností zůstávají některé získané schopnosti a životní zkušenosti zachovány až do smrti, stejně jako schopnost vnímat vnější svět a cítit pozitivní nebo negativní emoce.
Příčiny AN jsou velmi komplikované a pravděpodobně i poměrně variabilní. U tzv. familiární AN, která se objevuje v raném věku (často již před 60. rokem života), hrají velký význam genetické příčiny. Častější je však tzv. sporadická AN (85–90 % všech případů AN), jejíž příznaky se poprvé objevují typicky mezi 60. a 70. rokem života. Prvotním příčinám sporadické AN rozumí věda méně, roli ale hraje obecné opotřebení tkání (patologie lysozomů, dysfunkce mitochondrií, oxidativní stres či zánětlivé projevy).
Kritéria pro diagnózu AN stanovují dva hlavní dokumenty – DSM-5 (Diagnostic and Statistical Manual of Mental Disorders, edice 5) a NIA-AA (National Institute on Aging – Alzheimer’s Association Criteria). Hodnotí se přítomnost demence a nedostatečnosti v alespoň dvou kognitivních oblastech, pozvolný nástup a progresivní zhoršování stavu a dále je nutné vyloučit další možné příčiny demence. Diagnózu je možné podpořit také biochemickým vyšetřením mozkomíšního moku a mozkovými zobrazovacími metodami (PET, MR).
AN je v současnosti nevyléčitelná a používaná léčiva přináší pacientům jen poměrně malý prospěch. K léčbě se používají tzv. inhibitory acetylcholinesterázy (takrin, rivastigmin, galantamin a donepezil) a memantin ze skupiny inhibitorů NMDA receptorů. Kauzální terapie, která by umožnila ovlivnit ukládání beta-amyloidu a tau-proteinu do mozkové tkáně, dosud neexistuje. Výzkum léků zaměřený na amyloidovou cestu přináší slibné výsledky, takže stále existuje reálná naděje na léčbu, která bude prognózu pacientů s demencí zlepšovat (disease modifying drugs), v blízké době nejspíše v rámci biologické léčby. V klinické praxi mají i nadále nezastupitelnou roli psychosociální intervence a podávání kognitiv.
Frontotemporální lobární degenerace (FTLD) je klinicky i neuropatologicky heterogenní skupina neurodegenerativních onemocnění definovaných neuropatologickým podkladem s dominujícím postižením frontálního laloku a frontotemporálního pomezí (často ale bývají postiženy i parietální kůra a bazální ganglia). Neuropatologickým podkladem jsou intraneuronální depozita tau-proteinu (tauopatie) nebo jiných peptidů (non-tau FTLD), z nichž nejčastěji jde o protein TDP-43 (TAR DNA-binding protein 43; TDP-43 proteinopatie), ubikvitinu, proteinu FUS a dalších. Následně dochází k zániku postižených neuronů a sekundární astroglióze. FTLD se klinicky projevují ve třech možných kategoriích:
1/ behaviorální varianta FTD (bvFTD): dominuje změna osobnosti a chování, většinou provázená dysexekutivním syndromem;
2/ primární progresivní afázie (PPA): nově vzniklá a zpočátku izolovaná alterace řeči, která postupně progreduje do obrazu demence;
3/ kombinace demence a poruch hybnosti: extrapyramidové projevy u tauopatií – kortikobazální degenerace a progresivní supranukleární obrna; nebo onemocnění motorického neuronu u TDP-43 proteinopatií.
Na MR mozku je typicky asymetrická frontotemporální atrofie, častěji výraznější vlevo. Již v dřívějších stadiích bývá zřetelná obdobná distribuce hypoperfuze a hypometabolismu na SPECT, resp. PET mozku. Z hlediska patologického se jedná o tauopatie, ubikvitinopatie nebo demence bez specifického histologického obrazu. Pro diagnostiku pravděpodobné FTLD je nutno použít publikovaná klinická diagnostická kritéria. Léčebné možnosti jsou omezené.
Nově byla popsána také jednotka LATE (limbic-predominant age-related TDP-43 encephalopathy), která by mohla zahrnovat část klinických nálezů a nálezů na MR, jež jsou sice typické pro Alzheimerovu nemoc, ale jsou v rozporu s negativním výsledkem metabolických biomarkerů. Na základě současných teoretických modelů onemocnění je možné očekávat pozitivitu biomarkerů již 10–15 let před výskytem prvních klinických příznaků.
Progresivní supranukleární obrna je onemocnění z okruhu atypických parkinsonských syndromů řadící se mezi tauopatie. Mezi jeho hlavní příznaky patří poruchy okulomotoriky, časná posturální instabilita, symetrický hypokineticko-rigidní syndrom s axiální převahou a kognitivní deficit. Spektrum příznaků a rychlost progrese se liší v závislosti na konkrétní variantě onemocnění. Diagnostika se opírá především o klinický nález a z podpůrných metod zůstává na prvním místě MR. Supranukleární obrna je klasicky považována za sporadické onemocnění. Publikováno bylo pouze několik raritních případů s možným familiárním výskytem.
Kortikobazální degenerace je vzácné progresivní neurodegenerativní onemocnění ze skupiny tauopatií charakterizované parkinsonismem, kortikální atrofií četných oblastí mozku včetně mozkové kůry a bazálních ganglií, kognitivní dysfunkcí a postižením zrakově-prostorových funkcí. Začíná zpravidla ve věku nad 60 let a progreduje 5 až 10 let.
Vaskulární demence je heterogenní skupina onemocnění, která mají společného jmenovatele – kognitivní deficit na podkladě vaskulární etiologie, tedy jako důsledek ischemického postižení nebo hemoragického poškození mozkové tkáně.
Demence s Lewyho tělísky (DLB) je neurodegenerativní onemocnění na pomezí Parkinsonovy a Alzheimerovy nemoci. Má společné příznaky obou poruch a svoje vlastní specifické příznaky, především zrakové halucinace lidských postav a zvířat, fluktuující úroveň kognice a senzitivitu k neuroleptikům. Prevalence nemoci v pozdním stáří je zřejmě dost vysoká (odhad 10–20 % ze všech demencí). Klinicky se ji daří diagnostikovat málo často (4 %), nicméně diagnostický význam spočívá především v rozhodnutí o léčbě. Neuroleptika (antipsychotika) nejsou zpravidla lékem první volby. Jako vhodnější se jeví použití inhibitorů cholinesteráz nebo opatrné podávání nových antipsychotik s nepatrnou potencí k vyvolání parkinsonského syndromu.
Demence při Parkinsonově nemoci – onemocnění s prevalencí odhadovanou mezi 10 a 40 %. Kognitivní deficit nedosahující kritérií demence se vyskytuje pravděpodobně u více než poloviny nemocných PN. Stoupá s věkem a se stupněm motorického postižení. Vyšší prevalence je také u pacientů s pozdním začátkem onemocnění (nad 60 let věku) a s převažujícími akineticko-rigidními příznaky. Výskyt demence zhoršuje prognózu onemocnění. Demence je převážně podkorového typu. Progreduje porucha exekutivních funkcí, paměti a abstraktního myšlení.

Parkinsonova nemoc a další extrapyramidová onemocnění
Dle posledních odhadů postihuje přibližně 4,5 milionu osob ve věku 50 a více let v nejlidnatějších zemích, včetně západní Evropy. Důsledkem stárnutí populace se incidence a prevalence PN zvyšují a s postupujícím stavem onemocnění se zvyšují i motorické a autonomní příznaky a neuropsychiatrické komplikace. Prevalence PN se odhaduje zhruba na 0,3 % obecné populace a na 1 % u osob starších 60 let. Odhaduje se, že do roku 2040 se bude s PN léčit téměř 13 milionů lidí po celém světě. Onemocnění se obvykle začíná manifestovat během páté a šesté dekády, ale 10 % z celkového počtu pacientů začíná mít první obtíže již před 40. rokem věku.
Klinické příznaky se manifestují až při postižení 80 % dopaminergních neuronů substantia nigra. K tomu dojde mnoho let po spuštění vlastního patologického procesu, a proto lze klinické symptomy považovat za znak již pokročilého stadia onemocnění. Klinické projevy zahrnují motorické a nemotorické příznaky. Základním motorickým projevem onemocnění je parkinsonský syndrom, kombinace hypokineze s minimálně jedním dalším příznakem (rigidita, třes, porucha stoje a chůze). Mezi nonmotorické příznaky se již od časných stadií řadí vegetativní poruchy – zácpa a seborea. Z psychických změn je nejčastější deprese a izolované kognitivní deficity (zvláště exekutivní dysfunkce). Demence a polékové psychotické projevy se objevují až v pozdějších stadiích onemocnění, a to pouze u části nemocných.
Přítomnost parkinsonského syndromu je stěžejní pro diagnózu PN. Až 80 % pacientů s tímto syndromem trpí právě Parkinsonovou chorobou, u zbylých 20 % pacientů se pomocí anamnézy a dalších klinických příznaků zjišťuje jiná příčina. Diagnóza PN je stanovena na základě klinického vyšetření a anamnézy. Zobrazovací vyšetření jako magnetická rezonance a metody nukleární medicíny pomáhají pouze v případě klinicky nejasných stavů. Stanovení správné diagnózy je zcela nezbytné pro zahájení optimální terapie. Vzhledem k velmi pozvolnému nástupu a pomalé progresi onemocnění pacienti často přicházejí k vyšetření až ve fázi funkčního omezení. Na druhé straně zejména u mladých pacientů, u nichž začíná onemocnění před 40. rokem věku (young onset forma), dochází ke zpoždění stanovení diagnózy až o několik let. Důvodem je zejména skutečnost, že se na možnost PN v mladém věku dostatečně nemyslí, a navíc tito pacienti často nemají zcela typický průběh.
Onemocnění nemá kauzální léčbu. Základním substitučním lékem nadále zůstává levodopa. Agonisté dopaminových receptorů mají zpravidla nižší antiparkinsonský účinek než L‑DOPA, ale jejich užití snižuje výskyt pozdních hybných komplikací. Inhibitory katechol‑O‑methyltransferázy (entakapon, tolkapon) na periferii tlumí odbourávání L‑DOPA a zvyšují její množství procházející hematoencefalickou bariérou. Antagonista glutamátových receptorů s anticholinergním a dopaminergním účinkem (amantadin) a inhibitory MAO‑B (selegilin, rasagilin) jsou pomocnými léky s mírným efektem na projevy PN.
Pro dosažení významného zlepšení stavu u pokročilé PN jsou v současné době k dispozici pouze tři terapeutické možnosti: DBS (deep brain stimulation – DBS), kontinuální podkožní infuze apomorfinu a CDS – gel Duodopa (kontinuální duodenální dopaminergní stimulace Duodopou). Všechny tři možnosti jsou prokazatelně účinné a mají svoje výhody a nevýhody. Neurochirurgická terapie je indikována, když selhává farmakologická léčba. Z nefarmakologických strategií se osvědčila relaxace, podpůrná psychoterapie a postupy zahrnující tělesné cvičení.
PN je jedno z mála neurodegenerativních onemocnění, kde jsou k dispozici terapeutické možnosti, které výrazně snižují příznaky onemocnění. Pokud je léčba nasazena správně a pacient dodržuje doporučené postupy, je tíže hybných projevů minimalizována a kvalita života výrazně zlepšena.
Extrapyramidová onemocnění
Extrapyramidová onemocnění nejsou jednotně klasifikována podle jediné oficiální mezinárodní normy. Patří mezi ně atypické parkinsonské syndromy, esenciální tremor, Huntingtonova nemoc a další onemocnění, která se projevují poruchou hybnosti (třes, chorea, dystonie, myoklonus, tiky).

Epilepsie
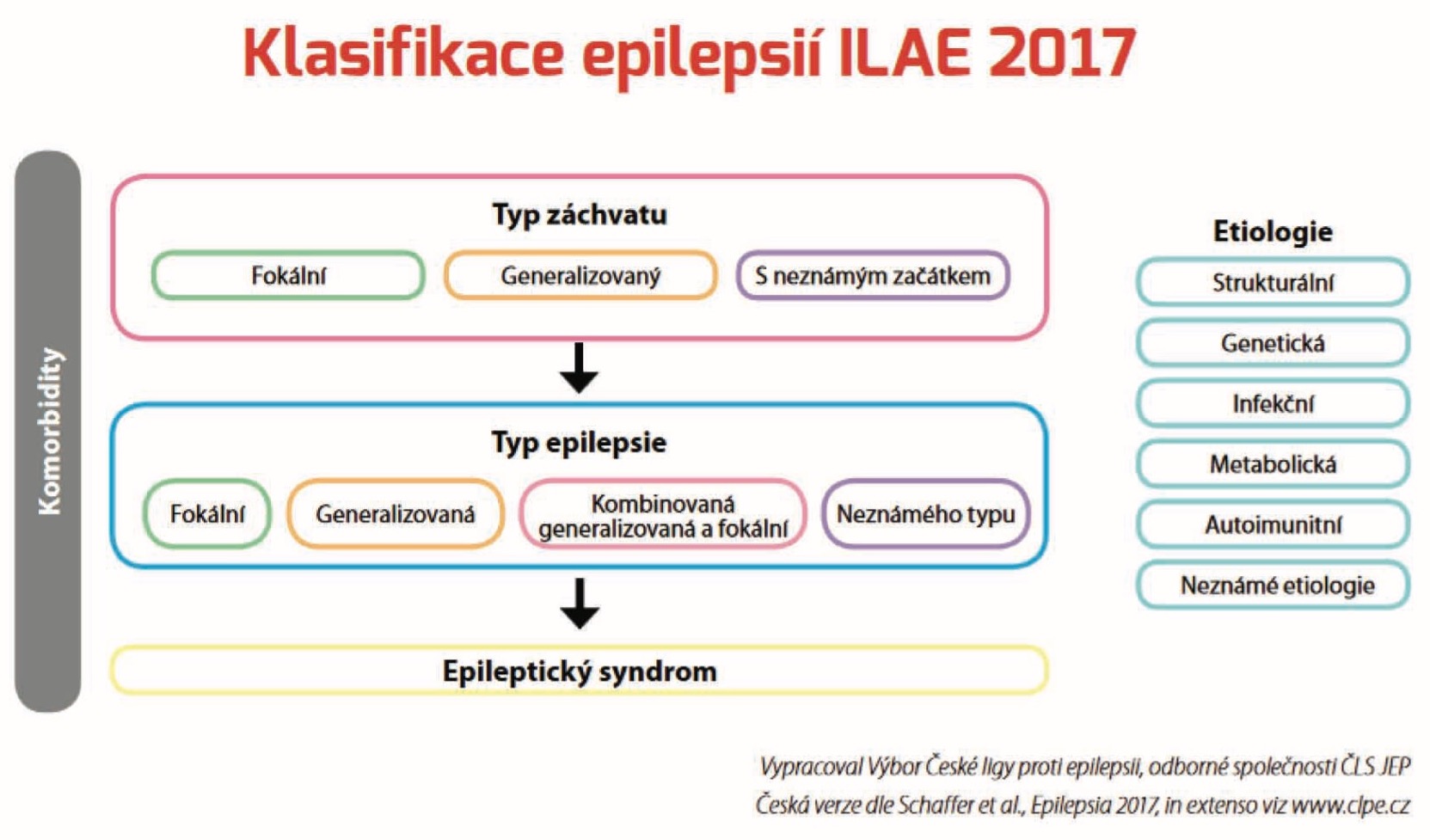
Dle současných kritérií je ke stanovení této diagnózy nutné, aby pacient prodělal dva epileptické záchvaty nebo jeden epileptický záchvat s vysokým rizikem jeho opakování. Epilepsií a epileptických syndromů je v současné době známo kolem 40 druhů. Manifestují se různými typy epileptických záchvatů a dalšími klinickými projevy. Mají odlišnou etiologii. Správné určení druhu epilepsie rozhoduje o terapeutickém postupu a prognóze.
Komise pro klasifikaci a terminologii Mezinárodní ligy proti epilepsii (ILAE) publikovala v r. 2017 dva klíčové dokumenty – novou klasifikaci epileptických záchvatů a novou klasifikaci epilepsií. Prvním krokem je klasifikace typu záchvatu. Klasifikace typu záchvatů i epilepsie zohledňují i výsledky vyšetření, jako jsou elektroencefalografie (EEG) a zobrazovací metody spolu s dalšími, která mají odhalit etiologii epilepsie. Druhou úrovní je určení typu epilepsie na základě stanovení diagnózy epilepsie podle definice z r. 2014.

 Při léčbě epileptických záchvatů se v první linii podávají benzodiazepiny. Chronická terapie je reprezentovaná 3 úrovněmi:
Při léčbě epileptických záchvatů se v první linii podávají benzodiazepiny. Chronická terapie je reprezentovaná 3 úrovněmi:
Úroveň 1 – režimová opatření, zejména dostatek pravidelného spánku a abstinence od alkoholu.
Úroveň 2 – chronická antiepileptická medikace, která je zahajována při splnění kritérií pro diagnózu epilepsie. V léčbě epilepsie dospělých stále dominuje farmakoterapie, která dokáže záchvaty kontrolovat u 70–80 % pacientů. Pro účinnou a bezpečnou léčbu epilepsií je vždy základem správná diagnóza. Podle současné klasifikace epilepsií připraví ošetřující lékař pro jedince s epilepsií, ať už nově diagnostikovanou, nebo aktivně probíhající, krátkodobý a dlouhodobý léčebný plán. Základní informace, jak v jednotlivých situacích postupovat, jsou uvedeny v souboru minimálních diagnostických a terapeutických standardů u pacientů s epilepsií, EpiStop 2021 (https://www.clpe.cz/soubory/epistandardy_2021_08_web--f792.pdf).
Úroveň 3 – chirurgická léčba epilepsie, která by se měla zvážit u pacientů, u nichž selhala dvě adekvátně zvolená, správně vytitrovaná antiepileptika bez ohledu na to, zda byla použita v monoterapii, či v kombinaci.
Chirurgickou léčbu epilepsie reprezentují dva typy výkonů – resekční a paliativní. Resekční výkony spočívají v odstranění části mozku, která je zodpovědná za vznik epileptických záchvatů. Výkony paliativní jsou v současnosti reprezentovány především neurostimulačními metodami.
Po jednom roce od implantace neurostimulátoru dochází přibližně u 40–50 % pacientů k 50% redukci epileptických záchvatů a toto číslo má tendenci v čase narůstat. Nicméně je nutné upozornit na fakt, že část pacientů (cca 25 %) na stimulaci nereaguje. Bohužel v současnosti není možné spolehlivě predikovat, kdo bude z implantace neurostimulátoru profitovat a kdo nikoliv.
Epilepsie a epileptické syndromy patří mezi nejčastější chronická neurologická onemocnění. Možnosti jejich diagnostiky a léčby se stále vyvíjejí a zdokonalují. Základním cílem je pacient bez záchvatů a bez nepřijatelných nežádoucích vedlejších účinků léčby, který může žít život podle svých schopností a předpokladů.

SMA a jiná nervosvalová onemocnění
V ČR by od 1. 1. 2022 měla být většina nemocných diagnostikována pomocí novorozeneckého screeningu, který byl nově rozšířen o vyšetření SMA. K diagnostice pak zbývají nemocní, kteří se narodili před zavedením screeningu, dále děti, jejichž rodiče screening odmítli nebo odmítnou, a pacienti s jiným typem SMA, než je SMA s mutací genu SMN1 (těch je přibližně 5 %). Základem správné diagnózy je pečlivý odběr anamnézy a objektivní neurologické vyšetření. Následují laboratorní odběry, kde může být zvýšená hladina kreatinkinázy (max. pětinásobek normy), a genetické vyšetření.
Formy SMA se různí podle věku pacienta v době prvních symptomů a nejvyššího dosaženého stupně motorických schopností při této diagnóze a počtu kopií SMN2. Forma 0, neonatální SMA, je málo častá, propuká již v děloze a většinou končí úmrtím do šestého měsíce života. Typ 1, infantilní SMA (m. Werdnig–Hoffmann), je nejčastější a tvoří 60 % nových případů. Typ 2, intermediální SMA, má očekávanou délku života bez léčby kratší než 2 roky. Typ 3, juvenilní SMA (m. Kugelberg–Welander), charakterizuje nástup prvních symptomů po 18 měsících věku. Nejméně častý je typ 4, adultní SMA, který se objevuje až v dospělém věku.
Typický průběh SMA neexistuje. Onemocnění probíhá u každého pacienta jinak a přechody mezi jednotlivými formami jsou plynulé. Z tohoto důvodu není možné dopředu předvídat průběh nemoci a léčbu je nutné přizpůsobit individuálnímu omezení funkčnosti a přítomnosti symptomů. Včasná diagnóza a zahájení terapie při dodržování souboru mezinárodně ověřených terapeutických kroků mohou průběh onemocnění a kvalitu života významně ovlivnit. SMA je onemocněním multiorgánovým, což by mělo být zohledněno při terapii.
V případě SMA je medicína svědky velikého přelomu v léčbě dosud neléčitelné a často fatální nemoci.
Nervosvalová onemocnění
Neuromuskulární onemocnění postihují periferní nervový systém. Jejich diagnostika včetně diferenciální diagnostiky je poměrně složitá. Důležitá je vždy pečlivá anamnéza a podrobné neurologické vyšetření, které klinikovi pomůže lokalizovat onemocnění do oblasti periferních nervů nebo svalů. Jedná se o velmi širokou problematiku, kam spadají například kompresivní nebo zánětlivé radikulopatie, ganglionopatie (léze ganglií spinálních či hlavových nervů), plexopatie, mononeuropatie či polyneuropatie. Dále se sem řadí celá problematika svalových onemocnění (vrozených či získaných), ale také poruchy neuromuskulárního přenosu (myastenie, LEMS apod.).
Ke stanovení diagnózy je vhodné provést laboratorní vyšetření krve (včetně glykemie, kreatinkinázy apod.) a mozkomíšního moku, poslat nemocného na elektromyografii, doplnit pomocné zobrazovací metody (rtg., CT, MR, sonografii apod.) a další vyšetřovací metody, které slouží k odlišení od jiných klinických stavů. U některých nemocných je vhodné doplnit biopsii svalu (včetně imunohistochemického vyšetření) a provést podrobné genetické vyšetření.
Po stanovení finální diagnózy je možné u řady chorob nervosvalové příznaky cíleně léčit (např. u Duchenneovy dystrofinopatie, spinální svalové atrofie nebo Pompeho nemoci). U autoimunitních poruch periferního nervového systému (příkladem jsou myasthenia gravis, autoimunitní neuropatie a zánětlivé autoimunitní myozitidy) lze využít více typů imunoterapie (imunosupresi, vysokodávkované imunoglobuliny, plazmaferézu, imunodifuzi a monoklonální autoprotilátky).

CMP
V souvislosti s iCMP se často lze setkat s pojmy minor stroke a tranzitorní ischemická ataka. Minor stroke představuje obecně malý, drobný iktus a nemá jednotnou definici. Bývá tak označována iCMP s lehkým neurologickým deficitem. Tranzitorní ischemická ataka (TIA) patří do skupiny iCMP se všemi možnými konsekvencemi včetně požadavku na urgentní diagnostiku a léčbu. Označuje se tak přechodný ložiskový neurologický deficit předpokládaného cévního původu, který odezní většinou do 1 hodiny, maximálně pak do 24 hodin.
CMP se projevuje náhle vzniklým ložiskovým neurologickým deficitem, který odpovídá teritoriu postižené mozkové tepny – nejčastěji jde o poruchu hybnosti a/nebo citlivosti poloviny obličeje, končetin nebo poloviny těla, poruchu řeči či dalších symbolických funkcí (např. apraxii), poruchu vizu, ataxii, deviaci hlavy a očních bulbů, pohledovou parézu, diplopii či náhle vzniklou nevysvětlitelnou závrať. Bolest hlavy, zvracení v úvodu, porucha vědomí nebo epileptické paroxysmy bývají přítomny vzácněji.
Péče o pacienty s CMP prodělala v ČR v posledních letech radikální změny díky dostupnosti nových léčebných postupů, jako je intravenózní trombolýza a endovaskulární terapie. Tyto postupy dnes představují standard léčby ischemické CMP vzniklé na podkladě uzávěru velké (proximální) mozkové tepny a patří mezi nejefektivnější léčebné metody současné medicíny.
Základní úroveň prevence iktu představují režimová opatření. Z populačního hlediska mají větší význam než prevence medikamentózní. Dodržování pravidel zdravého životního stylu ovlivňuje všechny známé (a jistě dosud nepoznané) ovlivnitelné rizikové faktory. Mezi nejdůležitější ovlivnitelné rizikové faktory patří arteriální hypertenze, dyslipidemie, diabetes mellitus 2. typu, kouření a přítomnost fibrilace síní. Mezi neovlivnitelné faktory patří věk, mužské pohlaví, pozitivní rodinná anamnéza a rasa (kvůli rozdílnému životnímu stylu i specifickému genetickému pozadí). Sekundární prevence iCMP by měla vycházet ze zjištěné etiologie a zahrnuje režimová opatření a farmakologické, chirurgické, endovaskulární a další specifické postupy. Základním specifickým farmakologickým postupem je terapie antitrombotická – antiagregační, antikoagulační či kombinovaná.
Antitrombotická léčba patří mezi pilíře farmakologických opatření prevence ischemického iktu. Možnosti jsou v zásadě dvě: použití antiagregační nebo antikoagulační terapie. Antiagregancia se podávají akutně při vzniku iktu i dlouhodobě u těch, kteří nejsou indikováni k léčbě antikoagulancii v kontextu velmi vysokého cévního rizika, jednoznačně pak při anamnéze aterotrombotické cévní příhody.
Antikoagulační léčba je dnes považována za samozřejmou součást prevence ischemické cévní mozkové příhody u pacientů s vyšším rizikem kardioembolického iktu při nevalvulární fibrilaci síní. Volí se v zásadě mezi warfarinem a newarfarinovými orálními antikoagulačními přípravky (NOAK).
Velice důležitá je komplexní následná péče o pacienta po iktu. I přes adekvátní intervence v primární a sekundární prevenci zůstávají následky CMP závažné. Do 3 měsíců po prodělané mozkové mrtvici 10–20 % pacientů umírá, 30–40 % nemocných zůstává trvale postižených a plné soběstačnosti dosáhne 50 % nemocných.

ALS
U ALS dochází k pozvolnému zániku periferních a centrálních motoneuronů a jejich drah s ušetřením extraokulárních a sfinkterových svalů. Rozlišuje se klasická forma ALS s postižením centrálního a periferního motoneuronu a dále progresivní bulbární paralýza s postižením bulbárních svalů. Vzácnější je progresivní svalová atrofie s lézí pouze periferního motoneuronu a primární laterální skleróza s postižením pouze centrálního motoneuronu. Existují formy ALS sdružené s demencí (frontotemporální demence, FTD-MND), u kterých jsou přítomné poruchy chování, kognitivní dysfunkce a postižení exekutivních funkcí.
Onemocnění je fatální a příčina není známa. Medián přežití jsou 2–4 roky. Kolem 50 % osob umírá do tří let od objevení se prvních příznaků, 90 % umírá do 5 let a jen kolem 5–10 % žije více než deset let od počátku choroby. Relativně delší přežití se objevuje u mladších osob a u těch, u kterých stanovení diagnózy trvalo delší dobu. Incidence v Evropě se uvádí kolem 1–2/100 000 obyvatel/rok, prevalence je 4–6/100 000 obyvatel.
U rozvinuté nemoci je klinický obraz poměrně charakteristický. Mohou se objevit bulbární příznaky s poruchou artikulace a obtížným polykáním. Často nemoc začíná asymetrickou slabostí ohraničené svalové skupiny, nejčastěji na horní končetině. V pletencových svalech horních a dolních končetin nebo na jazyku se objevují fascikulace či krampy. Pacient špatně chodí a má oslabené svaly horních a dolních končetin, zejména akrálně. S progresí nemoci se přidávají dechové obtíže.
Objektivní průkaz léze periferního motoneuronu v předních rozích míšních přinese elektromyografie (EMG), která má v diagnostice ALS klíčové místo. Nutné je také vyšetření mozkomíšního moku k vyloučení jiné etiologie (např. neuroboreliózy). Zobrazení mozku a míchy pomocí magnetické rezonance vyloučí další afekce nervového systému.
V současné době neexistuje na toto onemocnění specifický lék. Používá se neuroprotektivní léčba (např. riluzol), jejíž účinky nejsou jednoznačné, a léčba symptomatická, která je určena ke zvládání doprovodných projevů (dechových obtíží, hypersalivace, spasticity, depresí aj.). Důležitá je cílená a dlouhodobá rehabilitace a včasné požádání o vhodné pomůcky (vozík, polohovací postel, sedák do vany nebo na WC apod.).

Funkční poruchy hybnosti
FPH postihují především ženy (v poměru 2 : 1), nejčastěji ve věku 35–50 let, ale mohou se objevit i u dětí nebo seniorů. Jejich incidence se odhaduje na 80–140 případů na 100 000 obyvatel ročně. FPH se často vyskytují také v komorbiditě s jiným neurologickým onemocněním.
FPH nevznikají poškozením mozku, ale poruchou jeho fungování – například způsobu, jak mozek interpretuje vlastní pohyby a tělesné vjemy. Moderní neurobiologické teorie zdůrazňují roli tzv. prediktivního kódování – předpokládá se vznik abnormálních očekávání hybných a senzorických stavů zesílených abnormálně zaměřenou pozorností. Součástí patofyziologie je porucha vnímání volní kontroly pohybu. Na vzniku symptomů se mohou podílet i psychologické a sociální faktory, u všech pacientů však nejsou přítomny.
Diagnóza FPH se stanovuje na základě pozitivních známek inkonzistence (např. proměnlivosti příznaků), která je nekompatibilní s jiným neurologickým onemocněním, nikoliv jako diagnóza „per exclusionem“. Důležitou roli v diagnostice tak hraje neurolog, který pomocí specifických klinických testů zhodnotí přítomnost známek inkonzistence hybných projevů.
Léčba FPH vyžaduje komplexní individuální přístup. Základem je sdělení diagnózy s vysvětlením, že příznaky jsou skutečné a léčitelné. Klíčovou roli hraje specializovaná fyzioterapie, která obnovuje normální hybné vzorce a snižuje abnormální pozornost na pohyb. Nezbytnými součástmi péče jsou často psychoterapie a léčba komorbidit (např. depresí).
Prognóza závisí zejména na délce trvání potíží před stanovením diagnózy – časné rozpoznání a vhodná terapie výrazně zvyšují šanci na zlepšení.
